





系统:windows 11
时间:20260524
1. 软件的安装
1.1 傻瓜式安装
推荐的方式,相比于详细版后续又加入了许多新特性,后面也不打算维护详细版了。傻瓜式唯一需要动手的是安装WSL-Debian时候你的username和password,当然还有需要自己复制粘贴安装命令:
1.1.1 Install WSL (Windows)
Run in PowerShell or CMD on Windows:wsl --install -d Debian
1.1.2 Install Toolchain (Linux)
Run inside WSL Debian terminal (after installation):cd && \sudo apt update && \sudo apt install git -y && \rm -rf md-toolchain-wsl && \git clone https://github.com/sqzzfb/md-toolchain-wsl.git && \cd md-toolchain-wsl/linux && \chmod +x install_all.sh && \./install_all.sh && \cd ~ && \rm -rf md-toolchain-wsl && \mkdir calinout && \source ~/.bashrc
1.1.3 Other common used code
wsl -l -vwsl --shutdownwsl --terminate Debianwsl --unregister Debian
mda # Custom Keywordsjupyter notebook --no-browser
1.2 详细版安装
1.2.1 安装WSL2
有时候要更新WSL内核(CMD输入wsl --update或者去官网下载安装即可);在安装WSL之前,有时或许还要去“启用或关闭windows功能”开启所需要的功能(按win+R,输入optionalfeatures即可),,然后:
CMD或powershell输入:
wsl.exe --list --online # 查看可供直接安装的linux发行版
wsl.exe --install -d Debian # 安装喜欢的linux,默认是Ubuntu,本人比较喜欢Debian,故安装Debian
设置自己的username(以sq为例)和password,安装成功,按win键可以查看到新安装的WSL,点击WSL
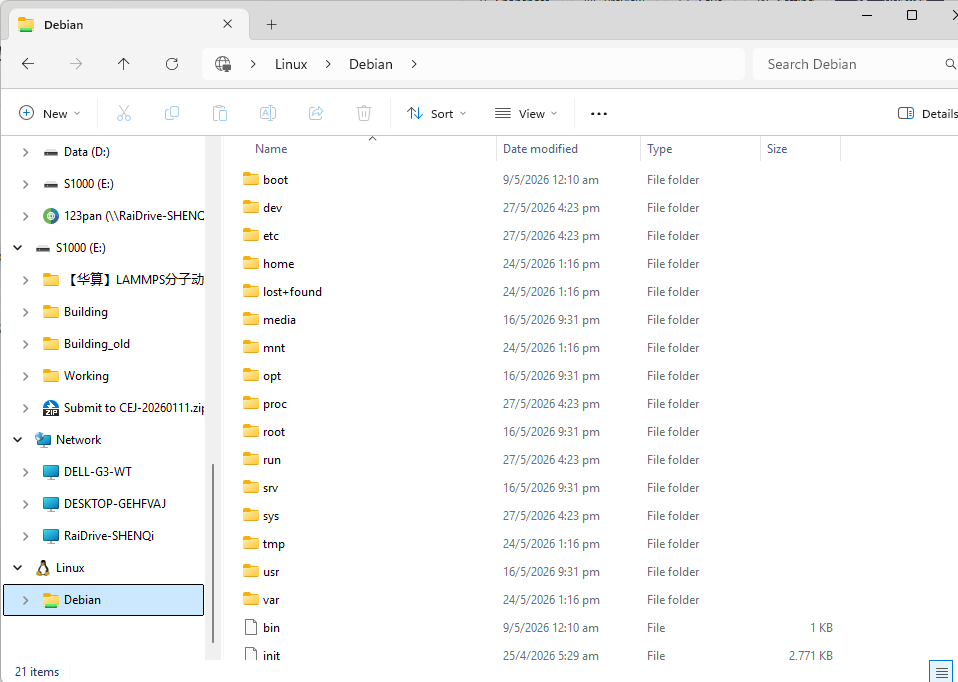
安装成功后去文件管理器,左侧栏会看到一个Linux小图标,点开会看到虚拟出来的Debian系统的文件夹(如下图)。后续如无必要,所有的文件都存放在这个虚拟分区中以便达到最高的运行效率
1.2.2 安装Packmol
确保退到用户文件夹
cd
更新,若需要password则输入刚刚设置的passwordsudo apt update
安装必要的工具sudo apt install -y gfortran make wget tar git
创建cal文件夹,用来安装软件程序,如果想换安装目录可以通过cd来实现mkdir cal
进入创建好的文件夹cd cal
下载最新源码包(如果由于网络问题下载慢,可以在浏览器下载后放到 WSL 里)wget https://github.com/m3g/packmol/archive/refs/heads/master.tar.gz
解压文件tar -xzvf master.tar.gz
进入解压后的目录cd packmol-master
编译Packmolmake
开始配置环境变量,以便在任何地方运行,打开 .bashrc 文件nano ~/.bashrc
滚动到文件最底部,添加以下内容(注意:将 /home/sq/cal/packmol-master 替换为你实际的绝对路径):export PATH="$PATH:$HOME/cal/packmol-master"
保存Ctrl + O
确认Enter
退出Ctrl + X
刷新环境变量,使配置生效source ~/.bashrc
验证安装packmol # 如果成功,会显示如下:
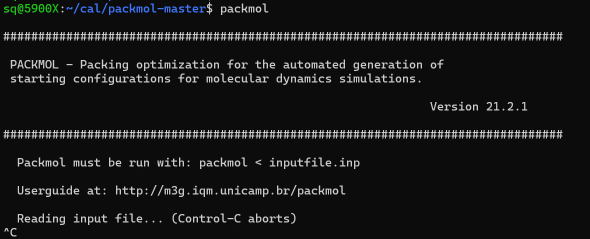
退出在运行的PackmolCtrl + C
1.2.3 安装Moltemplate
更新软件列表,升级系统里已经安装的软件,然后安装 Python 和依赖环境sudo apt updatesudo apt upgrade -ysudo apt install -y python3 python3-pip python3-venv python3-numpy python-is-python3
进到存放软件程序的文件夹cal
cd ~/cal
克隆仓库到本地,可能上不去网,要开下代理或者自己去手动下载git clone https://github.com/jewettaij/moltemplate.git
进入目录cd moltemplate
打开配置文件nano ~/.bashrc
在文件末尾添加以下三行,注意修改为你的实际路径,其中第3行非必需,作用是配置通用分子库路径,让 moltemplate 在任何目录下都能自动找到预设的力场和分子模型export PATH="$PATH:$HOME/sq/cal/moltemplate/moltemplate" export PATH="$PATH:$HOME/cal/moltemplate/moltemplate/scripts"export MOLTEMPLATE_PATH="$HOME/cal/moltemplate/moltemplate/common"
保存Ctrl + O
确认Enter
退出Ctrl + X
刷新环境变量,使路径生效source ~/.bashrc
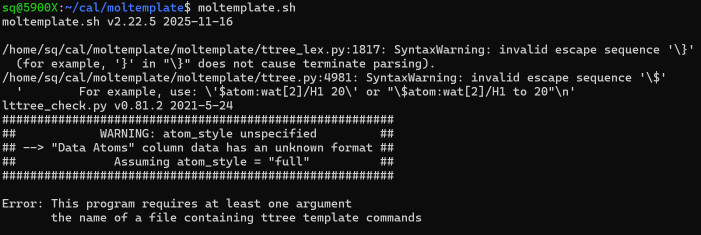
验证安装moltemplate.sh # 如果成功,会显示如下:
1.2.4 安装LAMMPS
进入自己设的安装计算软件的cal文件夹cd ~/cal
安装编译所需的依赖sudo apt updatesudo apt install -y cmake g++ openmpi-bin libopenmpi-dev libfftw3-dev libjpeg-dev libpng-dev
克隆LAMMPS 源码(使用浅克隆加速)git clone --depth=1 -b stable https://github.com/lammps/lammps.git
进入LAMMPS文件夹cd lammps
新建build文件夹,LAMMPS 采用经典的“脱产编译”(out-of-source build)方式,我们需要建一个专门的 build 文件夹mkdir build && cd build
使用 CMake 进行配置,这里我们默认开启几个分子动力学最常用的基础包:MOLECULE(分子),KSPACE(长程静电/PPPM),RIGID(刚体),MANYBODY(多体势如EAM),反应力场REAXFF等cmake ../cmake \
-D PKG_MOLECULE=yes \
-D PKG_KSPACE=yes \
-D PKG_RIGID=yes \
-D PKG_MANYBODY=yes \
-D DOWNLOAD_FFTW3=no \
-D BUILD_MPI=yes \
-D PKG_REAXFF=yes \
-D PKG_EXTRA-FIX=yes \
-D PKG_EXTRA-PAIR=yes \
-D PKG_MISC=yes
开始编译(-j $(nproc) 表示调用你 CPU 的所有核心加速编译)make -j $(nproc)
安装可执行文件make install
打开环境变量配置文件nano ~/.bashrc
配置环境,将make install得到的lmp文件所在的文件夹加入PATH,在最后一行输入export PATH="$PATH:$HOME/.local/bin"
保存Ctrl + O
确认Enter
退出Ctrl + X
刷新环境变量,使路径生效source ~/.bashrc
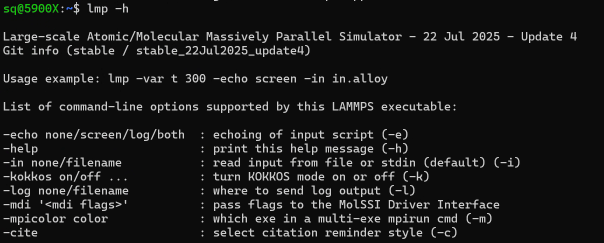
验证安装lmp -h # 如果成功,会显示如下:
后续扩展功能的方法
如果以后突然需要用到新功能(比如算石墨烯需要 AIREBO 势,需要开启 MANYBODY 之外的包,或者想算 GCMC 需要 MC 包):
重新回到build目录cd ~/cal/lammps/build
重新运行 cmake 命令,加上想要的新包(CMake 会自动保留你之前开启的包),以加一个 REPLICA 包为例cmake ../cmake -D PKG_REPLICA=yes
重新编译并安装,CMake 非常智能,它只会编译新加入的部分,几秒钟就能升级完成make -j $(nproc)make install
“较完整包”的安装方法
预编译版默认开启了非常多的包(几乎把能开的都开了)。如果担心自己编译版开的包以后不够用,想一步到位开启绝大多数常用包,可以在 CMake 阶段直接使用“全家桶”快捷命令(在 build 目录)。-D PRESET=most 是 LAMMPS 官方提供的一个预设组合,它会自动把 90% 的常用包(包括各类力场、分析工具)全部打包开启,BUILD_MPI=yes和PKG_REAXFF=yes是在此基础上加上“MPI并行计算”和“ReaxFF反应力场”cmake ../cmake \ -D PRESET=most \ -D BUILD_MPI=yes \ -D PKG_REAXFF=yes
重新编译安装make -j $(nproc) && make install
2. 开始模拟
2.1 AuToFF的使用
网站:https://cloud.hzwtech.com/web/personal-space/online-tool/auto-ff-home
选择需要的力场,如全原子力场,如图:
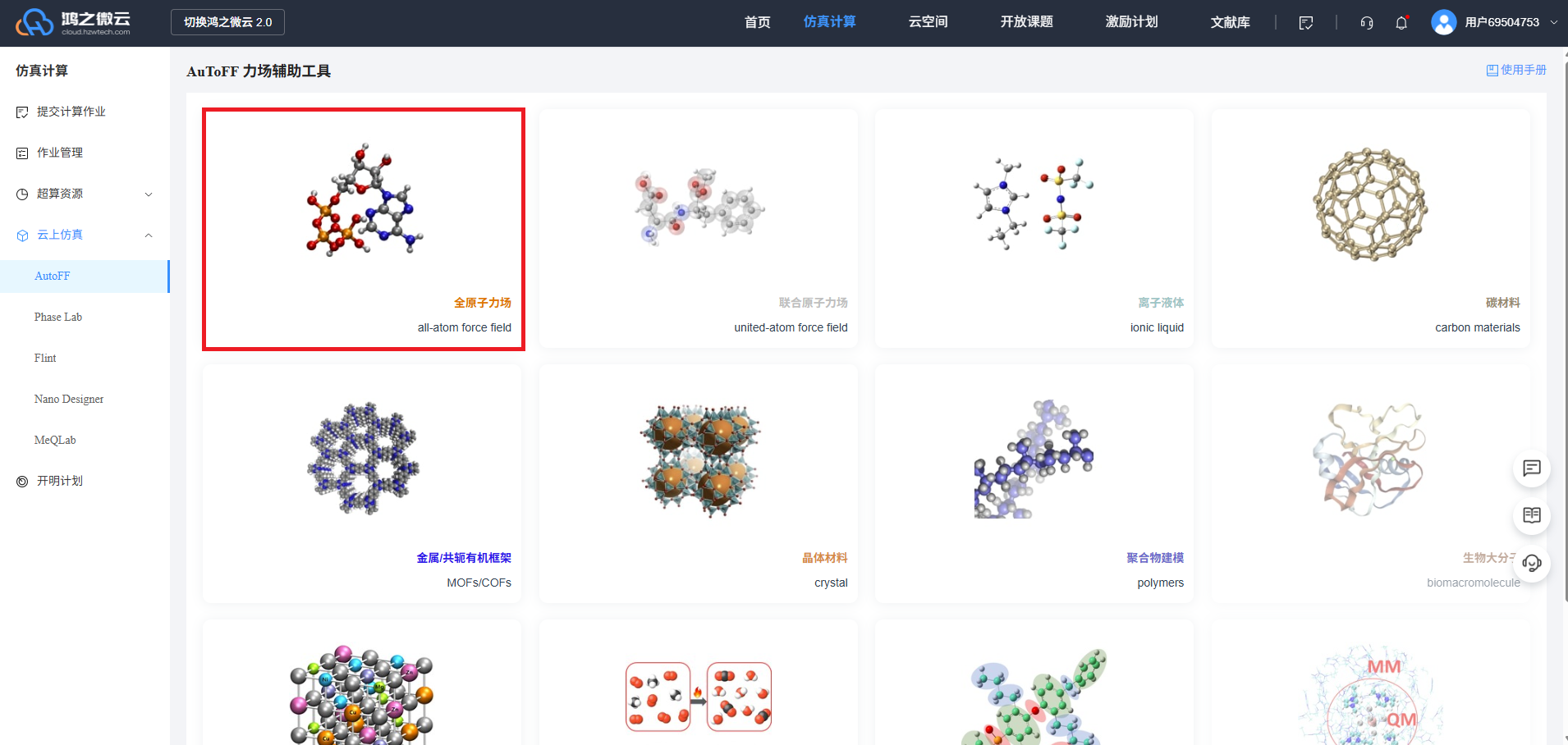
绘制用于建盒子的单个的分子,再点击生成3d结构视图,或者用materials studio等软件绘制后导出,再上传到AuToFF;
选择并拖动要用的力场,下一步,下一步,选择计算软件(moltemplate),点击下载,设置分子名称,OK,得到zip包;
类似的,操作得到所有分子的zip;
2.2 Moltemplate与Packmol的使用
解压得到所有分子的“lt”和“pdb”文件,还有重复的“system.lt”以及“packmol.inp”文件;
编辑“system.lt”和“packmol.inp”文件,以下为范例,注意“system.lt”和“packmol.inp”文件之间,分子顺序以及数量等要一一对应,但system.lt文件里面的盒子的大小要比“packmol.inp”的稍大一些,3Å即可,范例是5Å:
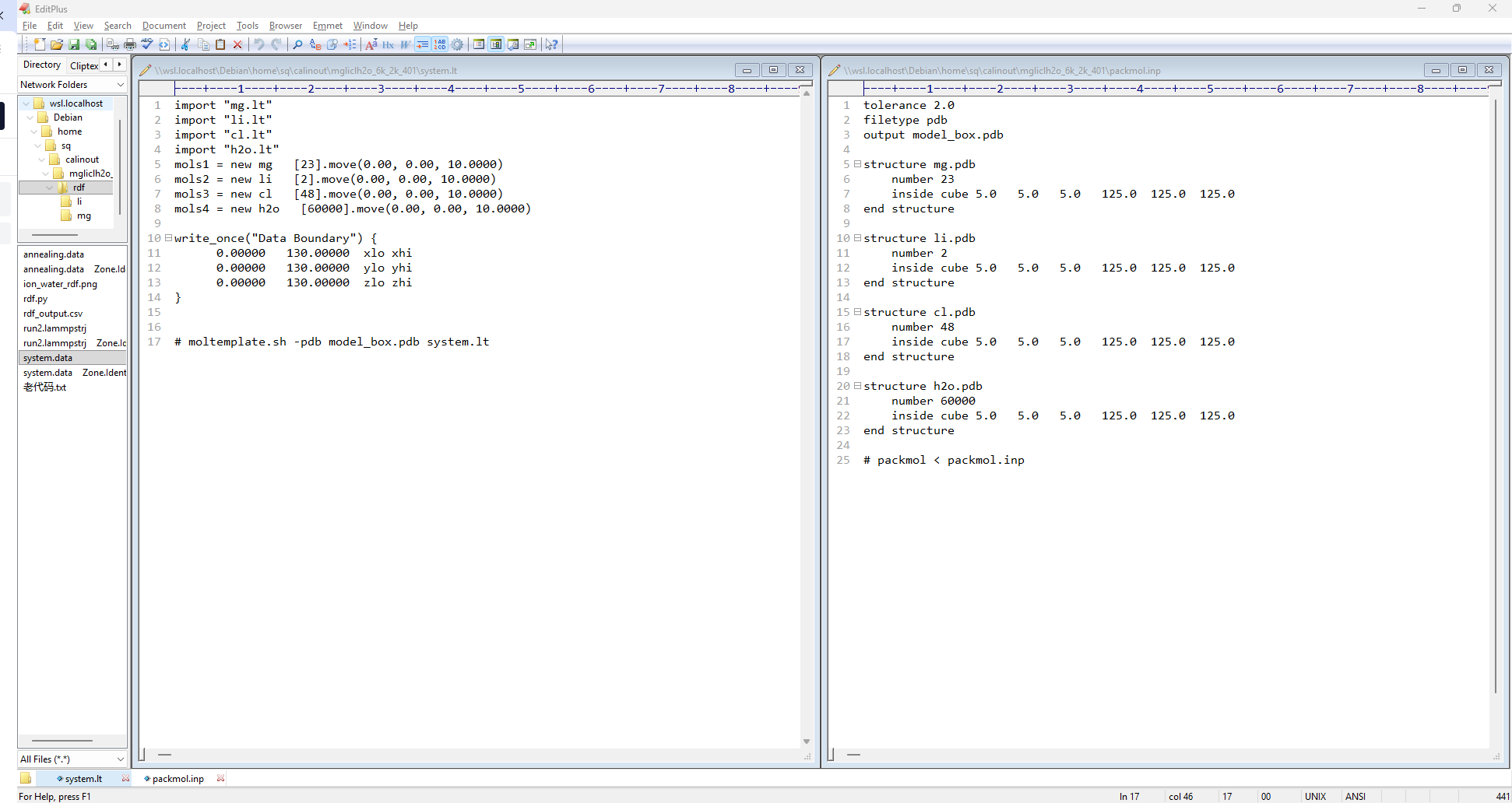
将所有分子的“lt”和“pdb”文件以及设置好的“system.lt”和“packmol.inp”文件放在同一个文件夹下,为计算整理方便,可以在/home/sq(你的用户名)路径下建立calinout文件夹,并在该文件夹下设立此次project名称的文件夹,将上述文件存放在此;
在该文件夹下按住shift,点击右键,open Linux shell here;
输入
packmol < packmol.inp,得到原子位置坐标信息盒子(output model_box.pdb文件);再输入
moltemplate.sh -pdb model_box.pdb system.lt为每个原子赋予刚刚在AuToFF设置的力场信息,得到的关键文件是“system.data”和“system.in.settings”,其中“system.data”文件为LAMMPS用到的data文件,包含原子之间的坐标和拓扑信息等,“system.in.settings”文件包含了对应的coeff等信息;
2.3 LAMMPS的使用
书写LAMMPS的in文件,以下为一个例子:
# mpirun -np 8 lmp -in Layer.in
units real # timestep 1 fs
boundary p p p
atom_style full
bond_style harmonic
angle_style harmonic
dihedral_style opls
improper_style cvff
pair_style lj/cut/coul/long 12
kspace_style pppm 1.0e-4 # ewald pppm
read_data system.data # TODO: your own data file
include system.in.settings # TODO: your own coeff file
thermo 100
thermo_style custom step density temp press ebond eangle lx ly lz
####### Minimization #######
velocity all create 300 730429
minimize 1e-8 1e-8 5000 5000
write_data min.data
print "Minimization Completed !"
####### Annealing #######
reset_timestep 0
dump 1 all custom 500 annealing1.lammpstrj id mol type x y z
dump 2 all custom 500 annealing2.lammpstrj id mol type xu yu zu
fix 1 all npt temp 300 300 100 iso 1 1 1000 drag 2 # Tdamp/Pdamp usually equal timestep*100/1000: 100=1fs*100, 1000=1fs*1000
run 20000
unfix 1
fix 1 all npt temp 300 400 100 iso 1 1 1000 drag 2 # Tdamp/Pdamp usually equal timestep*100/1000: 100=1fs*100, 1000=1fs*1000
run 10000
unfix 1
fix 1 all npt temp 400 400 100 iso 1 1 1000 drag 2 # Tdamp/Pdamp usually equal timestep*100/1000: 100=1fs*100, 1000=1fs*1000
run 10000
unfix 1
fix 1 all npt temp 400 300 100 iso 1 1 1000 drag 2 # Tdamp/Pdamp usually equal timestep*100/1000: 100=1fs*100, 1000=1fs*1000
run 10000
unfix 1
fix 1 all npt temp 300 300 100 iso 1 1 1000 drag 2 # Tdamp/Pdamp usually equal timestep*100/1000: 100=1fs*100, 1000=1fs*1000
run 20000
unfix 1
undump 1
undump 2
write_data annealing.data
print "Annealing Completed !"
####### Production Run #######
reset_timestep 0
dump 1 all custom 500 run1.lammpstrj id mol type x y z
dump 2 all custom 500 run2.lammpstrj id mol type xu yu zu
fix 1 all npt temp 300 300 100 iso 1 1 1000 drag 0 # if nvt: 1 all nvt temp 300 300 100 drag 0
run 50000
unfix 1
undump 1
undump 2
write_data final.data
print "ALL DONE !"将文件保存,我这里保存为Layer.in,即名称为Layer的in文件;
输入
mpirun -np 8 lmp -in Layer.in运行LAMMPS,模型就跑起来了,其中-np后面的8一般是你的电脑的核心数,更多细节不在这里阐述;如果有多个任务,不想浪费时间的话,可以设置排队运行,逻辑是使用
&&操作符,使得任务之间可以排队运行,避免上一个任务是凌晨结束,要到第二天早上才能续上任务的情况发生,节省时间,示范如下:mpirun -np 8 lmp -in Layer.in && cd /home/sq/calinout/mgliclh2o_6k_2k_401 && mpirun -np 8 lmp -in Layer.in,代码的含义是:现在当前文件夹下按照Layer.in运行LAMMPS,运行结束后跑去新的目录(mgliclh2o_6k_2k_401)下,按照新目录下的Layer.in文件运行新的LAMMPS工作。
2.4 MDAnalysis后处理数据
略,暂时停更




